

Posted by Dariia Dantseva, Lead Product Development Scientist on 26th May 2026
From $3 Billion to $599: The Complete History of Genome Sequencing
In 1953, James Watson and Francis Crick published the double helix structure of DNA in a one-page letter to Nature. The paper ended with one of the great understatements in scientific history: "It has not escaped our notice that the specific pairing we have postulated immediately suggests a possible copying mechanism for the genetic material."
What they had figured out was the shape of the molecule that carries all biological information. What they had not figured out - what would take another quarter century to solve - was how to read it.
The story of how science went from knowing DNA's structure to sequencing an entire human genome for under $600 is one of the more dramatic in modern biology. It involves two scientists racing independently to the same discovery, a $3 billion government project almost derailed by a private company, and a cost collapse so steep it makes the semiconductor industry look slow.

James Watson and Francis Crick with their DNA model at the Cavendish Laboratory in 1953.
The First Methods: Sanger and Gilbert (1977)
DNA sequencing - reading the actual sequence of nucleotide bases in a DNA molecule - was developed in 1977. Independently, simultaneously, and on opposite sides of the Atlantic.
Frederick Sanger at the MRC Laboratory of Molecular Biology in Cambridge developed what became known as chain-termination sequencing, or Sanger sequencing. His method works by copying a DNA template in the presence of chemically modified nucleotides - dideoxynucleotides - that terminate the growing chain at random positions. By running large numbers of these partial copies and separating them by size using gel electrophoresis, you can reconstruct the original sequence one base at a time.
At roughly the same time, Walter Gilbert at Harvard developed a chemical cleavage method that achieved the same result via a different route.
Both methods worked. Both were laborious. Both required significant technical skill and time.
Sanger and Gilbert shared the 1980 Nobel Prize in Chemistry - along with Paul Berg, who was recognised separately for his work on recombinant DNA. Neither man had collaborated with the other. They had simply been working on the same problem at the same moment, which happens in science more often than people expect.
Sanger sequencing became the dominant method for the next three decades. It was reliable, it was accurate, and by the standards of what came before it - which was essentially nothing - it was remarkable. It was also slow, expensive, and fundamentally limited in scale. Reading an entire genome one fragment at a time, by hand, was technically possible but practically inconceivable.

In 1980, Paul Berg, Walter Gilbert and Frederick Sanger were awarded a Nobel Prize for their contributions concerning the determination of base sequences in DNA.
A Stepping Stone: Direct Genome Sequencing (1984)
In 1984, a young postdoctoral researcher at UCSF named George Church co-authored a paper describing the first direct genome sequencing method - an approach that moved away from the gel-based readout of Sanger sequencing and toward a system that could be read electronically. It was an early step in a direction that would eventually become next-generation sequencing.
Church went on to become a professor at Harvard Medical School, co-found dozens of biotechnology companies, and become one of the most influential figures in modern genetics. He has been on The ODIN's scientific advisory board since the company's early days. His 1984 paper is a small piece of a very large story - but it was a real piece.
The Human Genome Project (1990–2003)
The Human Genome Project was announced in 1990. The goal was straightforward and enormous: sequence every base pair of a human genome - all 3.2 billion of them.
![]()
The project involved twenty institutions across six countries. It was funded by a combination of government agencies including the NIH, the US Department of Energy, and their counterparts in the UK, France, Germany, Japan, and China. The plan was to complete a reference genome in 15 years, at a projected cost of around $3 billion.
It nearly didn't happen that way.
In 1998, Craig Venter - then head of The Institute for Genomic Research - announced that his new private company, Celera Genomics, would sequence the human genome in three years using a different approach: whole-genome shotgun sequencing. Rather than systematically working through the genome region by region, Venter's method would break the entire genome into millions of small random fragments, sequence them all simultaneously, and use software to assemble the overlapping pieces into a complete sequence.
The public consortium dismissed the approach as unworkable for a genome as complex as a human's. Then Venter demonstrated it on a fruit fly. Then the race began.
Both efforts published their draft sequences within months of each other, in 2001. A joint announcement was made at the White House. The details of who actually finished first - and what "finished" meant - were disputed then and remain so now. The science benefited regardless.
The completed draft human genome was published in Nature and Science in February 2001. The cost of the public effort: approximately $3 billion. The time taken: 13 years.
What the genome contained surprised almost everyone. Rather than the anticipated 100,000 protein-coding genes, researchers found approximately 20,000 to 25,000 —-roughly the same number as a nematode worm. The vast majority of the genome was non-coding: regulatory regions, repetitive sequences, and stretches whose function remains incompletely understood to this day. The book of life turned out to be significantly stranger than the metaphor had implied.
Next-Generation Sequencing: The Cost Collapse (2005–present)
The Human Genome Project used Sanger sequencing at enormous scale. It worked, but it was not a sustainable model for sequencing many genomes. The cost and time required made routine whole-genome sequencing effectively impossible outside the largest research institutions.
Next-generation sequencing changed that equation completely.
NGS platforms - the first of which became commercially available around 2005 to 2007 - work by sequencing millions of DNA fragments simultaneously rather than one at a time. Where Sanger sequencing reads a single molecule, an NGS run reads hundreds of millions in parallel. The throughput increase was orders of magnitude. The cost implications were immediate.
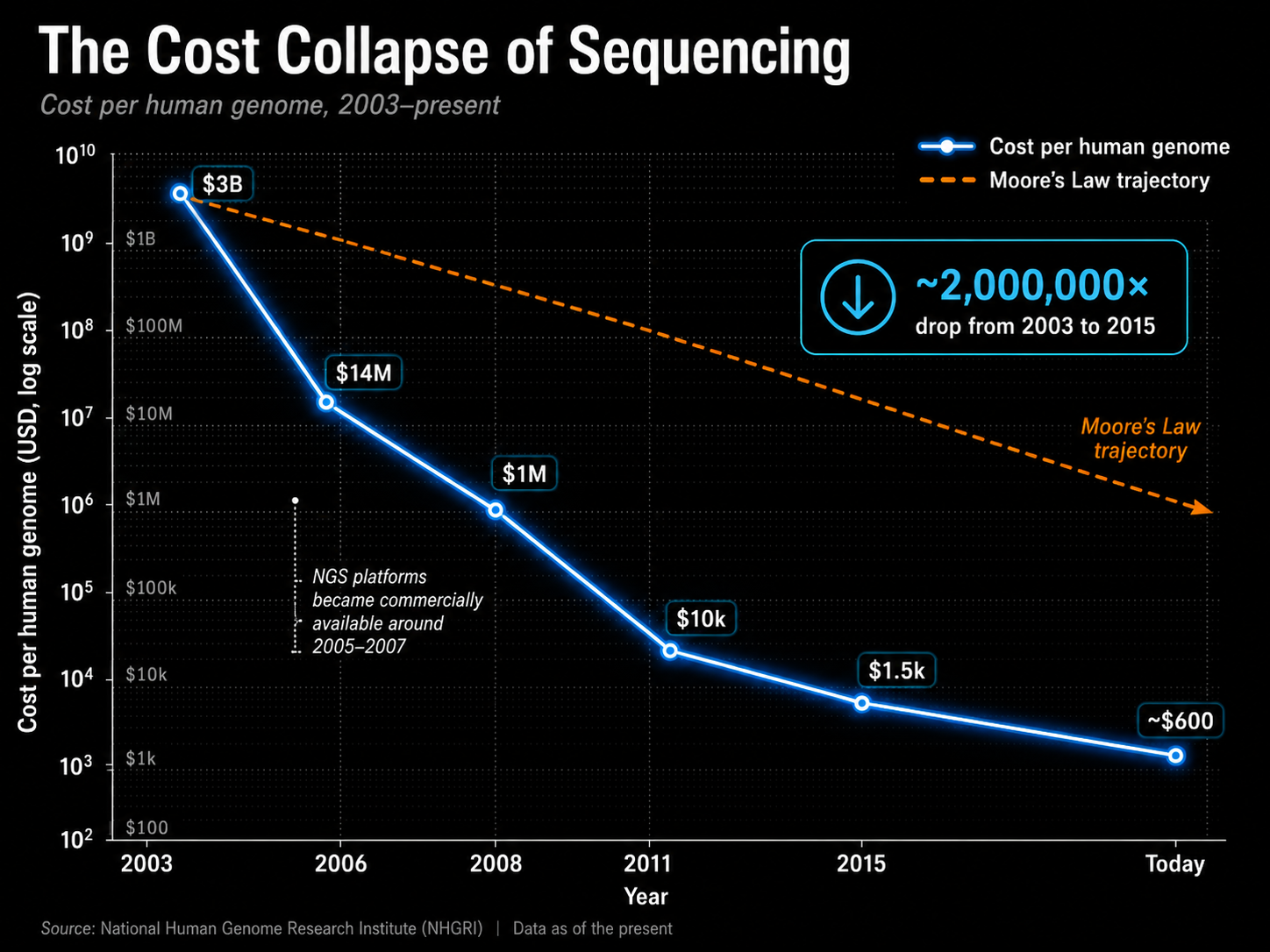
What followed was one of the steepest technology cost curves ever documented:
2003 — $3,000,000,000 (Human Genome Project)
2006 — $14,000,000
2008 — $1,000,000 2011 - $10,000
2015 — $1,500
Today — ~$600
For context: between 2003 and 2015 alone, the cost of sequencing a human genome fell by a factor of two million. Moore's Law - the benchmark for semiconductor progress, which roughly doubles computing power every two years - describes a doubling. The sequencing cost curve was exponentially steeper.
The National Human Genome Research Institute, which tracks sequencing costs, has noted that around 2007 to 2008, the curve dropped below Moore's Law trajectory and diverged dramatically downward. Sequencing got cheaper faster than almost any technology in history.
What Whole Genome Sequencing Actually Means Today
When someone offers whole genome sequencing today, the key metric to understand is coverage depth - expressed as a multiple like 10× or 30×. Coverage depth refers to how many times, on average, each position in the genome is read during sequencing.
30× coverage - the standard used in clinical research and hospital genetic testing - means every position in your genome is read approximately 30 times. Reading the same position multiple times allows errors to be identified and corrected statistically, producing a highly accurate final sequence.
The data you receive from 30× whole genome sequencing comes in three standard file formats:
FASTQ - the raw sequencing reads, exactly as the machine produced them. BAM - the reads aligned to a reference genome, showing where each fragment maps. VCF - the variant call file, identifying every position in your genome that differs from the reference.
These are the same file formats used by every sequencing facility in the world, from hospital genomics labs to pharmaceutical research departments to university sequencing cores. They're compatible with every standard analysis tool - IGV for visualisation, GATK for variant calling, PLINK for population genetics - because there is no alternative format at this level of the field.
Consumer DNA tests, by contrast, use a technology called SNP genotyping, which checks your genome at a set of pre-selected positions - typically around 600,000 - where variation is already known to exist. They cannot detect novel variants, structural changes, or anything in the regions between the positions they check. They are a different kind of product, answering different kinds of questions, from a different fraction of the genome.
The Access Problem
The sequencing technology is no longer the bottleneck.
Most whole genome sequencing still flows through institutional channels - university research programs, hospital clinical genetics departments, pharmaceutical company pipelines. These institutions have the compute infrastructure, the bioinformatics capacity, and the budget to handle the data. Individuals, independent researchers, small veterinary practices, and breeders generally do not.
That's been the default assumption underlying the field since the Human Genome Project: sequencing at this scale is institutional infrastructure. Not something an individual accesses directly.
That assumption is now outdated.
Whole Genome Sequencing at The ODIN
The ODIN offers 30× whole genome sequencing for $599.
A sample collection kit ships to you. Collection takes approximately five minutes. You mail the sample back with a prepaid label. In two to three weeks, you receive FASTQ, BAM, and VCF files - your complete genome sequence, in standard formats, ready for analysis with any tool you choose to use.
Your data is yours permanently. We don't sell it, share it, or retain it after delivery.
It works for any organism with a published reference genome - not just humans. Dogs, horses, cattle, zebrafish, salmon, Drosophila, and other model organisms are all supported. Breeders, veterinarians, conservationists, and independent researchers have all found uses the product page didn't anticipate.
George Church - who helped establish the technical foundations for modern genome sequencing in 1984 and has advised The ODIN since its early days - would probably describe this as the natural conclusion of a process that's been underway for forty years. The technology starts expensive and institutional. Then it becomes accessible.
That's where we are now.
Explore Whole Genome Sequencing at The ODIN →
Questions about whole genome sequencing, what it can and can't tell you, or which organisms are supported? Email us at odin@the-odin.com — we read and respond to these.