
This is by no means meant to be a comprehensive guide and is instead meant to show people the ease at which CRISPR-Cas9 can be used to modify the adult human genome.
CRISPR-Cas is a system with a terrible name that contrary to popular belief does not actually do any genetic engineering or modification of DNA bases directly. Instead, the system uses a trick that has been well known in genetic design for many many years. It cuts DNA. Yep, that’s all Cas9 does is cut DNA. See when DNA is damaged or cut most all organisms start to do DNA repair and this can end in one of three ways 1) The DNA is repaired perfectly and everything is ok 2) The DNA is repaired but some mistakes happen leading to problems translating the gene into protein because of frame shifts or mutations 3) The DNA is repaired using a template artificially supplied that results in a completely new sequence. When using CRISPR people aim to make use of (2) or (3) but most of the time when people talk about CRISPR they usually just mean (3) but not always.
Brief overview of how CRISPR-Cas9 works
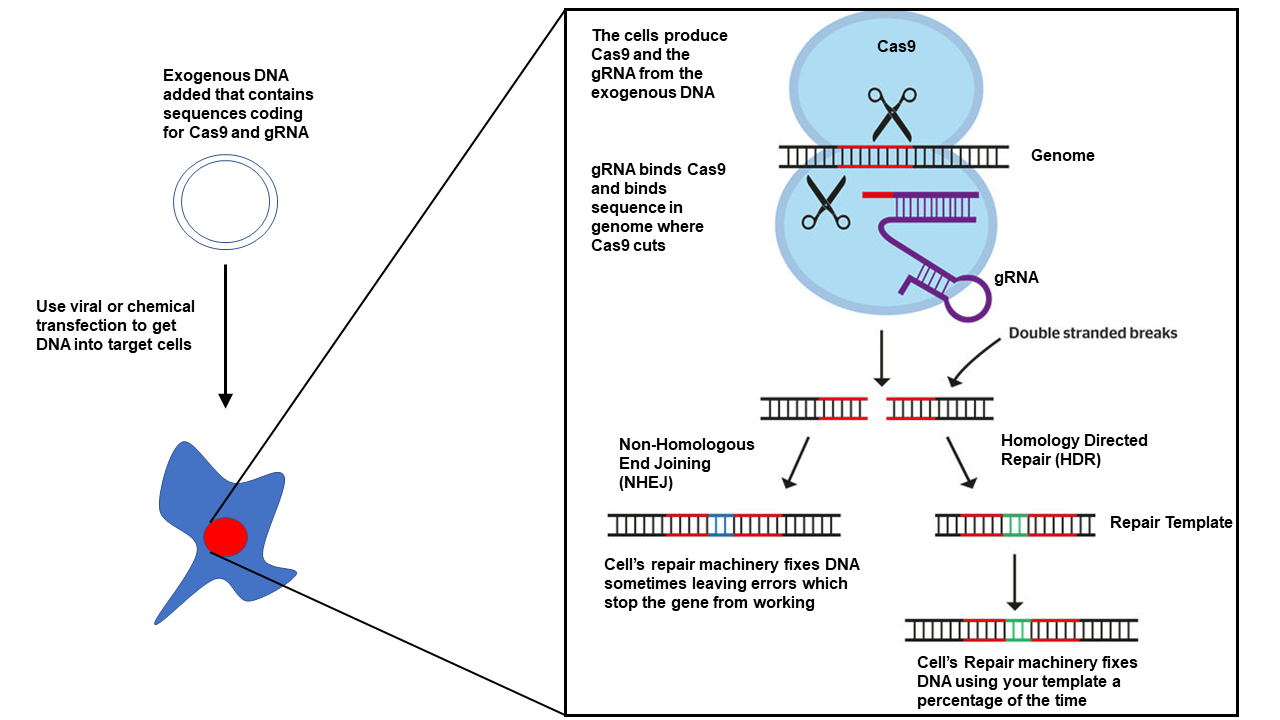
Most all CRISPR systems are composed of 2-3 components
-
The Cas9 protein which cuts the DNA
-
The tracrRNA and crRNA, which when synthetically combined are called a “guide RNA” but also called sgRNA(synthetic guide RNA) or gRNA
-
The template for repair if doing homology directed repair
For each CRISPR experiment you need to figure out before you start
-
Where in the genome will this happen?
-
What do I want to put into the genome or what base changes do I want to make?
TLDR; If doing a gene knock-out
Choose your guideRNA that will target your gene of interest using one of these websites and then order from Addgene or Atum.
Buy an endo-free maxiprep kit and purify your DNA from bacteria or pay one of a number of places to make the DNA for you
Mix with polyethylenimine(PEI) at a ratio of about 1μg DNA to 10μg PEI and inject >20μg of DNA. Injections will most likely need to be done multiple times to engineer enough cells to have an effect.
Step1: Which Cas9 should I use?
There are more Cas9s out there then seasons of Survivor which is still running(one of my favorite shows). SpCas9, SaCas9, nickase Cas9, Cpf1, dCas9…. The list goes on. Addgene wrote up brief discussion of some of the Cas9s if you are interested. However, in most cases it is easiest to use the wildtype SpCas9. While there might be so called “off-target” effects. The efficiency, accessibility and usability you have with it are better than the rest. Most systems that you order pre-made will come with wildtype spCas9 and so this makes your work much much less.
Step 2: Where do you want to make your change?
This usually comes down to two things
-
Do you want to modify a specific gene?
-
Do you just want to insert something in the genome and it doesn’t matter where?
Specific Genes
When using CRISPR Cas9 mediated genome modification you will either modify an existing gene or insert something new. When modifying an existing gene the sequence of the gene can be obtained from one of the many databases that hold human genome information. Many companies now have programs that can automatically target most genes so all you really need to know is the name of the gene that you want to target.
You can search and obtain sequences of genes at NCBI: https://www.ncbi.nlm.nih.gov/gene
New Gene
If your goal is to insert something in the genome of the organism and it doesn’t matter where there are what you can consider standard places that people insert things in organism genomes. These regions have usually been tested to show little or no effect on the organism.
These regions are Human Rosa26 or H11 ortholog locus or AASV1 More information about these and other sites can be found at : http://www.bushmanlab.org/assets/pdf/publications/22129804.pdf
Depending on how serious your experiment is you might want to sequence these regions to look for organism specific mutations but in most cases you can probably just use the sequences available from NCBI.
The reason we obtain these sequences is that they allow us to do two things
-
Design a gRNA to insert something in the genome
-
If doing homologous recombination, create homology arms for our template sequence so it is inserted into the genome
Step 3: Designing Your Guide RNA
One of the massive problems with CRISPR is the terminology. You don’t actually design the gRNA. You only design ~20 bases of the >90 base gRNA. This ~20 bp region contains two elements you should know about, one called the protospacer or sometimes just “spacer” and one called the PAM or Protospacer Adjacent Motif. Geesh I know. The PAM is needed in the genome for the gRNA to bind but is not needed in the gRNA.
If you want to try and be hardcore you can design gRNAs by hand but the great thing is that many companies and organizations have built up platforms and infrastructure so that designing gRNAs or (proto)spacer is automated.
Addgene has a list of validated gRNAs in a number of organisms including humans that you can diectly order.
https://www.addgene.org/crispr/reference/grna-sequence/
My favorite is Atum who easily allows you to design a gRNA either to a custom sequence or a gene found in their database. It costs ~$300 for a gRNA in a plasmid that also expresses Cas9. i.e. it is ready for for use in humans.
https://www.atum.bio/eCommerce/cas9/
ChopChop is great and gives many more targets than others and much more information
Remember, when I say we are designing a guide RNA we are really only designing 20 base pairs of the guide RNA known as the (proto)spacer. The rest of the guide RNA will almost always stay the same. The (proto)spacer is also located in the crRNA for those working with crRNAs
The (proto)spacer should be 20 bases long after and should match the genome where the cut will be made the one caveat is that you can’t match anywhere only places where there is an NGG(“N” means any nucleotide) sequence. So to design your gRNA what you do is find NGG in the top strand(what we have) and use the 20 nucleotides preceding it as your spacer.
Example
Partial Sequence Homo sapiens myostatin (MSTN), RefSeqGene (LRG_200) on chromosome 2
4801 agatttattt cttttatgaa gtagtcaaat gaatcagctc acccttgact gtaacaaaat
4861 actgcttggt gacttgggac agacagggtt ttaacctctg acagcgagat tcattgtgga
4921 gcaagagcca atcatagatc ctgacgacac ttgtctcatc taagttggaa tataaaaagc
4981 cacttggaat acagtataaa agattcactg gtgtggcaag ttgtctctca gactgtacat
5041 gcattaaaat tttgcttggc attactcaaa agcaaaagaa aagtaaaagg aagaaacaag
5101 aacaagaaaa aagattatat tgattttaaa atcatgcaaa aactgcaact ctgtgtttat
5161 atttacctgt ttatgctgat tgttgctggt ccagtggatc taaatgagaa cagtgagcaa
5221 aaagaaaatg tggaaaaaga ggggctgtgt aatgcatgta cttggagaca aaacactaaa
5281 tcttcaagaa tagaagccat taagatacaa atcctcagta aacttcgtct ggaaacagct
5341 cctaacatca gcaaagatgt tataagacaa cttttaccca aagctcctcc actccgggaa
5401 ctgattgatc agtatgatgt ccagagggat gacagcagcg atggctcttt ggaagatgac
5461 gattatcacg ctacaacgga aacaatcatt accatgccta cagagtgtaa gtagtcctat
5521 tagtgtatat caacagttct gctgactgtt gttctagtgt ttatgagaaa cagatctatt
5581 ttcaggctct tttaaacaag ctgttggcct gtatgtaagt agaaaggaaa agagtttctc
5641 tttttcaaga ttgcatgaga atatattaat gagacaaaaa tctgctgcat tatttgtttt
5701 cttatagaga caaaaaacta aaaaataaag tacttgcata gcattaattt aataaggcaa
5761 atatagatag catgcttatg ctttcacaat aataccacca aggcaaggac tgggagatac
gRNA (proto)spacer
TGACAGCAGCGATGGCTCTT
Total gRNA as DNA
TGACAGCAGCGATGGCTCTTGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGC
Looking at the information on NCBI we can find out that these sequence is in the first exon. It is usually best to target the first exon when using NHEJ(Non-Homologous End Joining) to knock out the function of a gene. You can then proceed to compare this to the whole human genome to see if it matches other places using BLAST. However, just use one of the links above those websites will do all of this for you.
Step 3b(optional): Designing Your Repair Template
The repair template is used when you are not just trying to knock-out a gene and instead are trying to actually make a specific change to the genome or add something in. As you can imagine this process is less efficient as it requires extra processes. However, this is the most promising use of CRISPR-Cas9 as there are many fewer processes that can be helped by simply knocking out a gene.
In order for the repair template to work it needs to have regions that match the genome. Basically, what you are trying to do is trick the cell into thinking that the repair template you are flooding the cell with is actually just a legit copy of the genome. It’s not and that’s why this work.
To do this you need what are called Homology Arms. These are regions that match the genome on the outside that flank our gene to insert. The word homology means descent with ancestry. So these arms are descendants of the genome essentially. We are basically making a fake genomic region.
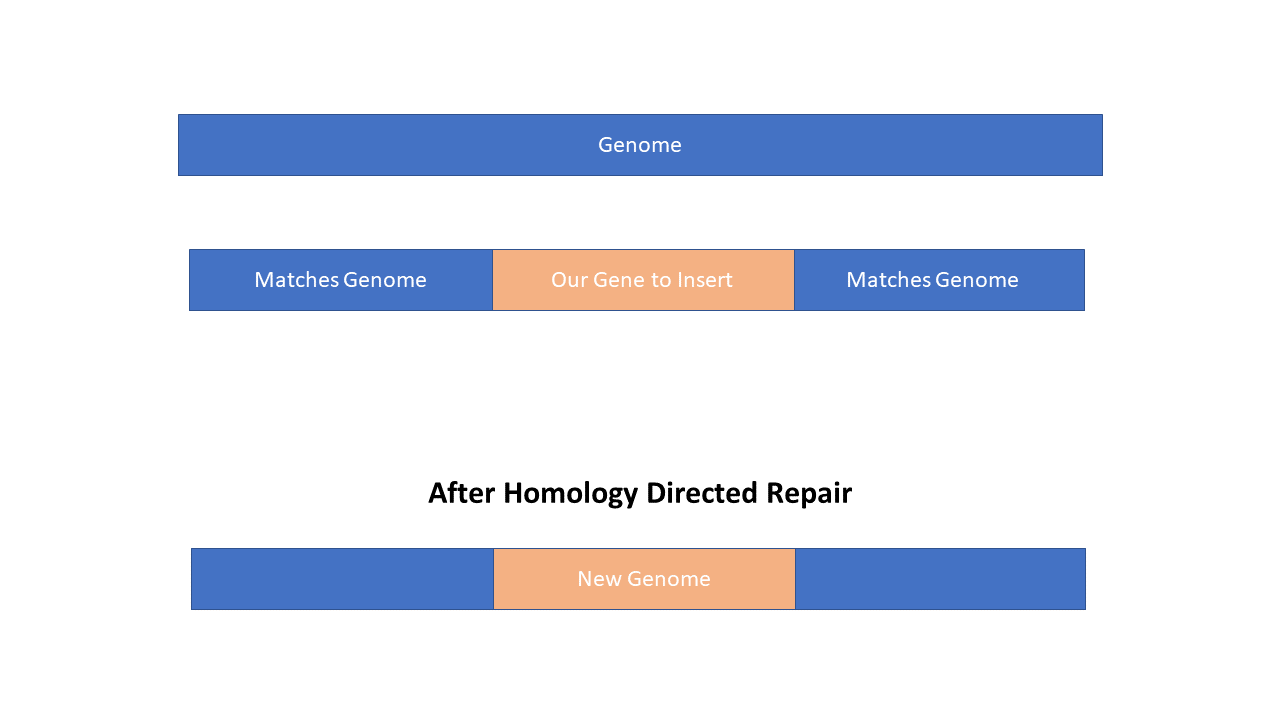
Now the size of the Homology Arms are important. If they are too small and the gene you are trying to insert is too big we won’t be able to fool the cell that our template is real. So if you are trying to insert a gene you need Homology Arms on each side of 800bpish which is pretty big! If you are only making single point mutations or changing one or two bases you can go as low as 50-100bp. The Homology Arms work best when they are within 100bp of the cut site near the gRNA but even better if they are within 10bp. Buttttt if you include the (proto)spacer and NGG(CCN) in your template beware because the CRISPR-Cas9 system will also cut your template if you are using double stranded DNA because it matches it also!!! The trick is to change the NGG in your template so that the gRNA won’t match it.
Step 4: Getting CRISPR-Cas9 into your Cells
There are three main ways to get the CRISPR Cas9 system into your cells
-
Adeno Associated Viruses(AAV)
-
Lentiviruses
-
DNA Transfection
Efficiency or transfection efficiency refers to the ability of the DNA to make it inside your cells. Better efficiency means more of your cells can undergo CRISPR reactions.
AAV
AAVs are one of the safest and easiest way to get DNA into your cells. They are fast becoming the method of choice to get DNA into cells for gene therapy. However, they have two limitations that make them difficult to use for CRISPR-Cas9. The first is that AAVs are limited in the amount of DNA they can hold. Cas9 is a pretty big protein and usually takes up a large portion of the AAV so that one cannot usually do HDR with a single AAV. What usually happens is that Cas9 is put in one AAV and the guide and template in another AAV. Needing two viruses to infect cells decreases the efficiency alot as cells need to be infected by both viruses so that HDR can occur. The second problem is that it is not easy to produce viruses as they require growing up cells that can be infected with the virus(mammalian cells) which are difficult to grow up in large quantities so viruses can be size, cost and material prohibitive especially for someone working at home. However, there are many companies that will make and sell you custom AAVs. Understand, these AAVs are replication deficient so they don’t reproduce they only infect cells and release your custom DNA. Search online you can find plenty.
Lentivirus
Lentiviruses have some pros and cons compared to AAVs. They have bigger genomic size so you can put alot more into them and only need one virus for an HDR CRISPR-Cas9 experiment. But they also integrate into the genome which can be dangerous as it increases the possibility that the genetic engineering or gene therapy you are doing will have something go wrong. Like AAVs these aren’t as DIY friendly but you can definitely find places to make custom lentiviruses for you.
DNA Transfection
DNA transfection is a general term used to describe getting artificial produced DNA(usually replicated in bacteria) and introduce this DNA into cells using one of a variety of methods, including chemical transformation or electroporation. I am a sucker for transfection because the DNA can be replicated in bacteria which can be grown on basically some sugar water. It can be purified for human usage in most any basic lab setup. All you really need is a centrifuge and some pipettes. What it lacks in efficiency as compared to viruses can be made up for in quantity. You can purify alot easily so even if the efficiency is lower you can use more. This DNA can literally be applied directly to the skin(though very lower efficiency), can be injected with the DNA in water, higher efficiency), injected with the DNA mixed with a chemical or lipid(even higher efficiency and best choice) or injected and then stimulated with an electrical current(highest efficiency but more impractical). The best compound to use based on efficiency, price and ability to obtain is probably polyethylenimine(PEI).
You can find a Human Myostatin targetting knock-out CRISPR plasmid using the gRNA above in our store. It is not injectable and not meant for human use