
You can find the plasmid DNA sequences
CRISPR is a system with a terrible name that contrary to popular belief does not actually do any genetic engineering or modification of DNA bases. Instead, CRISPR uses a trick that has been well known in genetic design for many many years. It cuts DNA. Yep, that’s all a CRISPR system does is cut DNA. See when DNA is damaged or cut most all organisms start to do DNA repair and this can end in one of three ways 1) The DNA is repaired perfectly and everything is ok 2) The DNA is repaired but some mistakes happen leading to problems translating the gene into protein because of frame shifts or mutations 3) The DNA is repaired using a template artificially supplied that results in a completely new sequence. When using CRISPR people aim to make use of (2) or (3) but usually just (3).
This means that most all CRISPR systems are composed of 3 components
-
The Cas9 protein which cuts the DNA
-
The tracrRNA and crRNA, which when synthetically combined are called a “guide RNA” but also called sgRNA or gRNA
-
The template for repair
There are two unknowns then for every CRISPR experiment that you need to figure out
-
Where in the genome will this happen
-
What do I want to put into the genome or what base changes do I want to make
Further considerations include
-
How will I select for my change(do I need to include selection markers in my gene insertion)?
-
If bacteria, am I using a lab strain that lacks the repair machinery? For most cloning strains(DH5a, DH10B) the answer will be yes. If so they will not work with good probabilities with CRISPR
-
If bacteria, is the strain of bacteria I am using already resistant to antibiotics? (Many lab strains can be(DH10B is streptomycin resistant, HME62 is ampicillin resistant, some BL21(DE3) strains are Tet resistant, &c) then you can’t use these antibiotics as selection markers
Where do you want to make your change?
This usually comes down to two things
-
Do you just want to insert something in the genome and it doesn’t matter where?
-
Do you want to modify a specific gene
If your goal is to insert something in the genome of the organism and it doesn’t matter where there are what you can consider standard places that people insert things in organism genomes. These regions have usually been tested to show little or no effect on the organism.
Bacterial region between the ybhH and ybhD genes
Yeast
Plants
Drosophila
Mouse Rosa26 or H11 locus
Humans Rosa26 or H11 ortholog locus or see http://www.bushmanlab.org/assets/pdf/publications/22129804.pdf
Depending on how serious your experiment is you might want to sequence these regions to look for organism specific mutations but in most cases you can probably just use the sequences available from NCBI.
To find the sequences start here https://www.ncbi.nlm.nih.gov/genome/browse/
After searching for your organism and the results are returned Click the first link at the top next to “Reference Genome” then scroll down to the bottom and you should see this
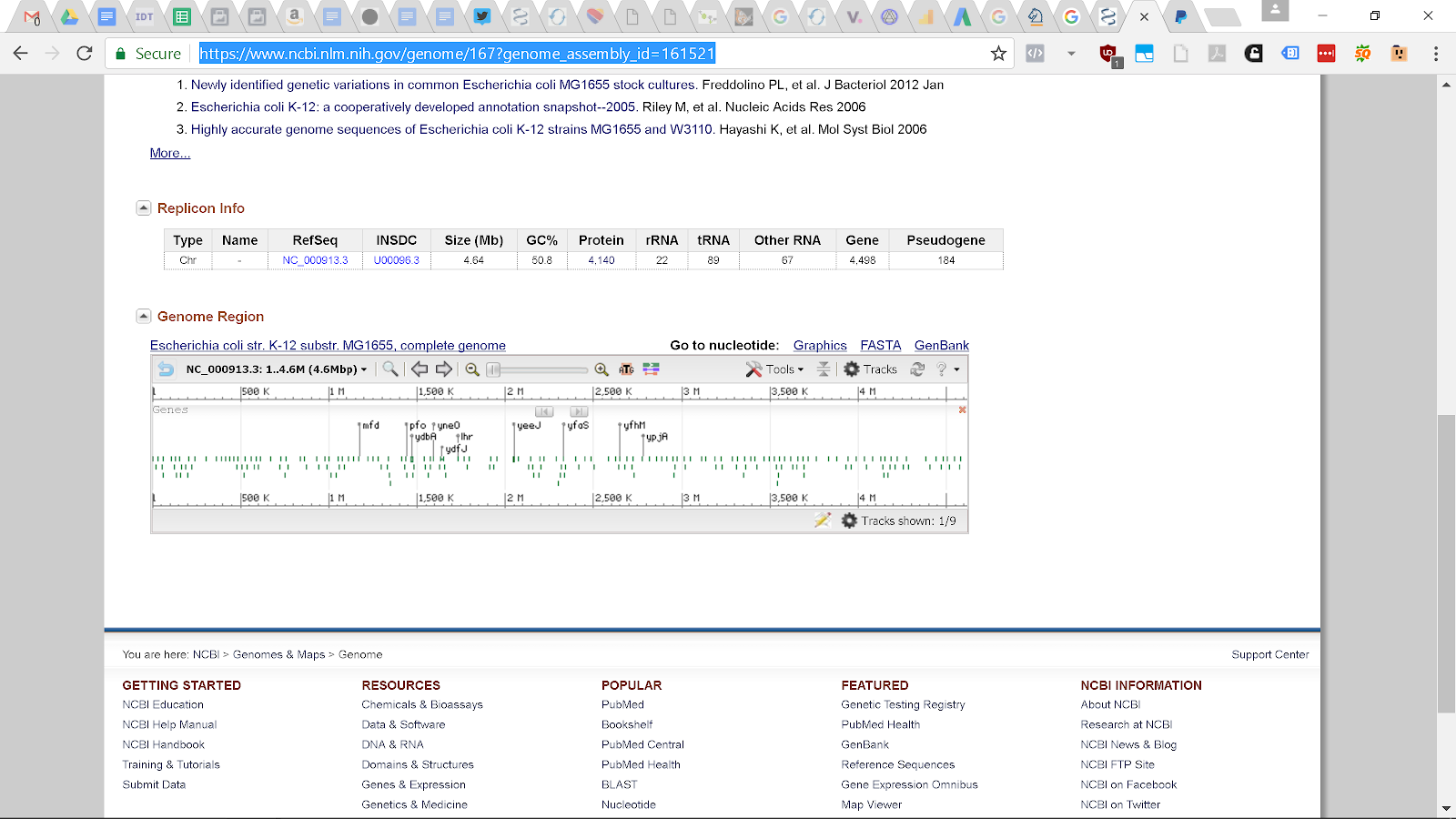
Click on the “Tools” icon and if you know the name of the gene, like “ybhD” for bacteria, you can search for it. The gene after ybhD is ybhH and the region between them is where we want to insert our gene. How do I know this? From experience. You can also search for ybhH and find out where it is located and deduce from there. From literature people have inserted between these genes with no adverse effects to the bacteria.
Next we want to get the sequence between these genes so we can create our guide RNA and our template DNA. Or if you are hardcore, design primers and sequence this region for better specificity.
If you zoom in and put your pointer over the ybhD gene you see that it starts at base number 800,575 and you can see that ybhH starts at 800,759. So we can search again using the term 800575..800759 then highlight a region and using your mouse over the highlight region select “Modify Range” put in the range 800575..800759 then hover your mouse over again and select “Download FASTA Sequence” then open the sequence file using notepad and it should contain
>gi|556503834|ref|NC_000913.3|:800575-800759 Escherichia coli str. K-12 substr. MG1655
TTATTACTCCGGAAAATGGAAGCGACGATTTTGGGTGGCTGGCCGTTAAAAATTTTAACTGCATTTAGCCAACTTAAATTAATGAAAAAATGTTATTAATCGTTGAGCTAAAGTCATTAGAGATGCTTTGCCCTTAATGTAACCATATCGCAATAAGTTATGTTTTTAAATTGAGGGCATTATTA
This sequence allows us to do two things
-
Design a guide RNA to insert something in the E. coli genome between ybhD and ybhH
-
Create homology arms for our template sequence so it is inserted into the genome
When I say we are designing a guide RNA we are really only designing 20 base pairs of the guide RNA known as the spacer. The rest of the guide RNA will almost always stay the same. The spacer is also located in the crRNA for those working with crRNAs
The spacer should be 20 bases long and it should end in CCN(“N” means any nucleotide) or NGG, this is called the PAM or Protospacer Adjacent Motif. What you do is find NGG in the top strand(what we have) and use the 20 nucleotides preceding it as your spacer or find CCN on your top strand and use the reverse complement of 20 nucleotides after it as your spacer, which will make the CCN, NGG on the strand that the spacer is bound to.
For many organisms you can use a design tool to select the best spacer at https://www.atum.bio/eCommerce/cas9/results?multipleContacts=false
Scanning through the ybhH-ybhD region we can identify a number of targets let’s pick something in the middle
TTATTACTCCGGAAAATGGAAGCGACGATTTTGGGTGGCTGGCCGTTAAAAATTTTAACTGCATTTAGCCAACTTAAATTAATGAAAAAATGTTATTAATCGTTGAGCTAAAGTCATTAGAGATGCTTTGCCCTTAATGTAACCATATCGCAATAAGTTATGTTTTTAAATTGAGGGCATTATTA
Using the design tool we can choose a spacer
ATGGAAGCGACGATTTTGGG
Next we should pick the homology arms. Homology arms are regions in your template that are identical to the genome of the organism you are trying to insert something into. This makes it so that the organism actually thinks your template is a real template. These arms go on each side of the template DNA and the size depends on the species you are trying to insert something into and the size of what you are trying to insert into the genome.
For bacteria and yeast homology arms on each side of 40bp-100bp work pretty well if you are inserting a few kb of DNA. If you are only making single point mutations or changing one or two bases you can go as low as 20bp-30bp. The homology arms work best when there are within 100bp of the cut site but even better if they are within 10bp. If you include the spacer and NGG(CCN) PAM beware because the CRISPR system will also cut your template!!!
For us the upstream homology arm can be 40bp and have this sequence
TTTTGGGTGGCTGGCCGTTAAAAATTTTAACTGCATTTAG
And the downstream homology arm can be 40bp and have this sequence
ACTTAAATTAATGAAAAAATGTTATTAATCGTTGAGCTAA
If you have the gRNA plasmid from our CRISPR kit then you can add in this new spacer pretty easy.
You can do a rolling circle PCR amplification to change the DNA sequence
Forward primer
GTCCTAGGTATAATACTAGT + Spacer + GTTTTAGAGCTAGAAATAGC
Reverse Primer
The reverse primer should just be the Reverse complement of the Forward Primer
GCTATTTCTAGCTCTAAAAC + Spacer + ACTAGTATTATACCTAGGAC
So our primers for this example would be
Forward Primer ATGGAAGCGACGATTTTGGG
GTCCTAGGTATAATACTAGT + ATGGAAGCGACGATTTTGGG + GTTTTAGAGCTAGAAATAGC
Reverse Primer
GCTATTTCTAGCTCTAAAAC + CCCAAAATCGTCGCTTCCAT + ACTAGTATTATACCTAGGAC
gRNA plasmid Fwd Sequencing Primer
GGCTGGCTTAACTATGCGGC
PCR - use proofreading polymerase pfu
35 cycle
94C - 30s
55C - 30s
68C - 3 minutes
After the PCR do a DpnI digest to remove the old plasmid by adding 1uL of DpnI to the PCR reaction and incubate at 37C for 1 hour. Do a cleanup using a column if you have miniprep or cleanup columns.
Transform into Dh5a or DH10B and select with Amp
Purify and miniprep the plasmid and send off for sequencing using the gRNA Fwd sequencing primer.
To create the template you need to do PCR
To amplify GFP with promoter and terminator from pJK prok1 eGFP plasmid
Forward Primer with homology arm
TTTTGGGTGGCTGGCCGTTAAAAATTTTAACTGCATTTAGCACAGCTAACACCACGTCGT
Reverse Primer with homology arm
TTAGCTCAACGATTAATAACATTTTTTCATTAATTTAAGT CAAAAAACCCCTCAAGACCC
PCR - use proofreading polymerase pfu
30 cycle
94C - 30s
56C - 30s
68C - 1 minute 30 seconds
Do a DpnI digest by adding 1uL DpnI and then incubating at 37C for 1 hour and then do a PCR purification
At this point in time you should be able to do a transformation into a recbombinase positive strain of E. coli or transform using our Cas9 plasmid + arabinose as it contains recombinases in the plasmid.
Transform with Cas9 containing plasmid and the gRNA containing plasmid and the GFP template and select with Kanamycin and Ampicillin and look for fluorescent colonies.