

Posted by Dariia Dantseva on 22nd Apr 2026
What is whole genome sequencing — and what can it tell you about yourself?
Your genome contains approximately 3 billion base pairs of DNA. Packed inside every cell in your body, it's the most complete biological record of who you are — and for most of human history, it was completely unreadable.
That changed. And now, for $599, you can have yours sequenced in full.
Whole genome sequencing (WGS) has crossed a threshold. What once required institutional funding, specialized infrastructure, and years of waiting is now accessible to individuals, breeders, researchers, veterinarians, and agricultural producers. If you or your organism has a reference genome, you can get a complete genetic readout — raw data included — in 2–3 weeks.
Here's what it actually means, and what you can do with it.
What is whole genome sequencing — and how is it different from other DNA tests?
Most DNA tests you've encountered — ancestry kits, breed panels, disease screening arrays — look at a pre-selected list of positions in the genome. They're designed around known variants: things science has already characterized and decided are worth testing for. That makes them fast and cheap. It also means they can only tell you what they were built to find.
Whole genome sequencing doesn't have that constraint. It reads every base pair across the entire genome — not a curated subset, not targeted regions. The complete sequence. If a variant exists anywhere in the genome, WGS will find it. That includes known variants covered by consumer panels, but also rare alleles, novel mutations, structural variations, and anything else that might be clinically or scientifically meaningful — now or in the future.
That last part matters: because you receive the raw data, your genome can be reanalyzed as science advances. A variant that isn't understood today might be well-characterized in five years. With a WGS file on hand, you don't need to sequence again.
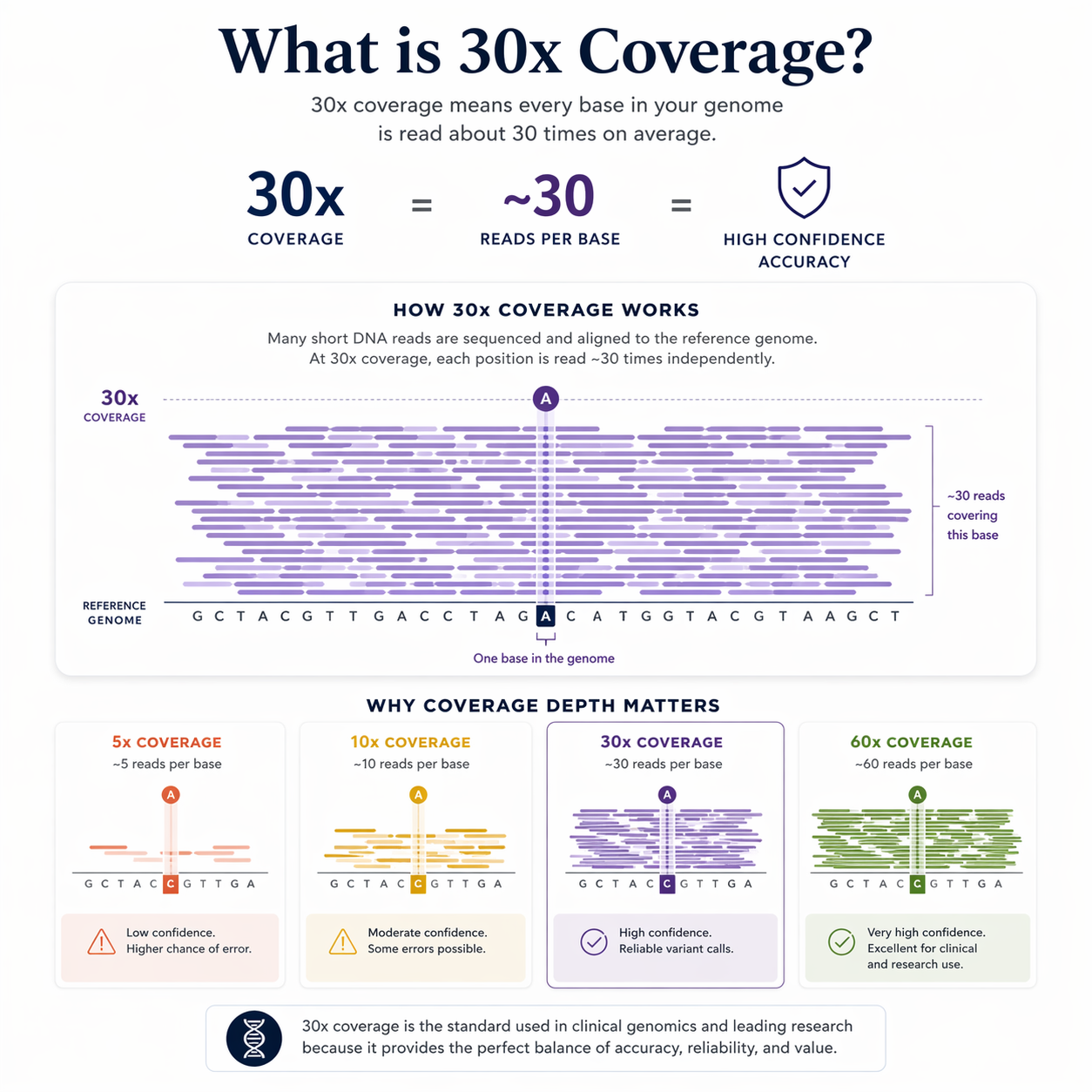
What does "30x coverage" mean — and why does it matter?
Coverage depth is how many times each position in the genome is independently read during sequencing. At 30x, every base pair has been read approximately 30 times.
This matters because sequencing isn't perfect at the individual read level. A single read might contain an error. Reading the same position 30 times independently and reaching a consensus is what gives you confidence that a variant call is real — not an artifact of sequencing noise.
30x is the threshold used in clinical human genomics, rigorous research applications, and elite breeding programs. Below 10x, variant calls become unreliable. At 30x, you're working with data you can act on.
Our sequencing runs on Illumina PE150 — paired-end reads of 150 base pairs from both ends of each DNA fragment. Paired-end sequencing improves alignment accuracy across the genome, especially in repetitive regions, and produces more reliable variant calls as a result.
What do you actually receive?
Every order includes three core data files in industry-standard formats:
Raw sequencing data (FASTQ). The foundational output of the sequencer — billions of short reads, each with an associated quality score. This is your complete, unprocessed genetic record.
Aligned reads (BAM file). Your raw reads, mapped to the reference genome for your species. This is what you use to visualize coverage, inspect specific regions, and identify structural variants.
Variant call file (VCF). A standardized file listing every position where your organism differs from the reference — SNPs, insertions, deletions, structural variants. This is where the biologically meaningful information is concentrated.
These files work with the full ecosystem of bioinformatics tools: IGV, GATK, PLINK, Samtools, bcftools, and most research pipelines. Whether you run your own analysis or share the files with a researcher or clinician, you're working with formats that integrate seamlessly into existing workflows.
What can you do with your genome data?
For individuals: Whole genome sequencing gives you a permanent, complete genetic record. It can inform conversations with clinicians about disease risk, support rare disease investigation, provide the deepest possible picture of ancestry and population genetics, and serve as a baseline for longitudinal health tracking. Because you own the raw files, you can share them with researchers, run them through emerging interpretation tools, or simply hold them for future use as genomic science matures.
For breeders: Whether you work with horses, dogs, cattle, or any other species, WGS gives you a complete picture of your animals' genetics — not just the variants on a standard panel. Use it to screen for hereditary conditions, assess genetic diversity within your program, make pairing decisions based on actual genomic data, and build genetic health records that add documented value when buying or selling.
For veterinarians and clinicians: WGS can clarify an unexplained phenotype, confirm a suspected genetic condition, or provide the genomic baseline needed for long-term health management. When a panel test comes back negative but the clinical picture doesn't fit, a whole genome sequence is often where the answer lives.
For researchers: Generate publication-quality WGS data for population genetics, comparative genomics, variant discovery, or association studies. The FASTQ, BAM, and VCF files slot directly into standard research pipelines.
For agricultural producers: Accelerate selective breeding programs, identify production-relevant alleles, reduce the generational time needed to improve a herd or crop line, and build genomic databases that compound in value over time.
Which organisms can be sequenced?
Any organism with a published reference genome. That includes:
- Humans — personal genomics, health, research, ancestry
- Sport and companion animals — horses, dogs, cats
- Livestock and production animals — cattle, sheep, pigs, goats, poultry
- Aquaculture species — Atlantic salmon, tilapia, sea bream, and others
- Agricultural crops and plants — wheat, maize, soybean, and more
- Research model organisms — mouse, zebrafish, Drosophila, and others
- Wildlife and conservation species — with appropriate sample collection protocols
Reference genome databases are growing rapidly. If you're unsure whether your species qualifies, reach out before ordering.
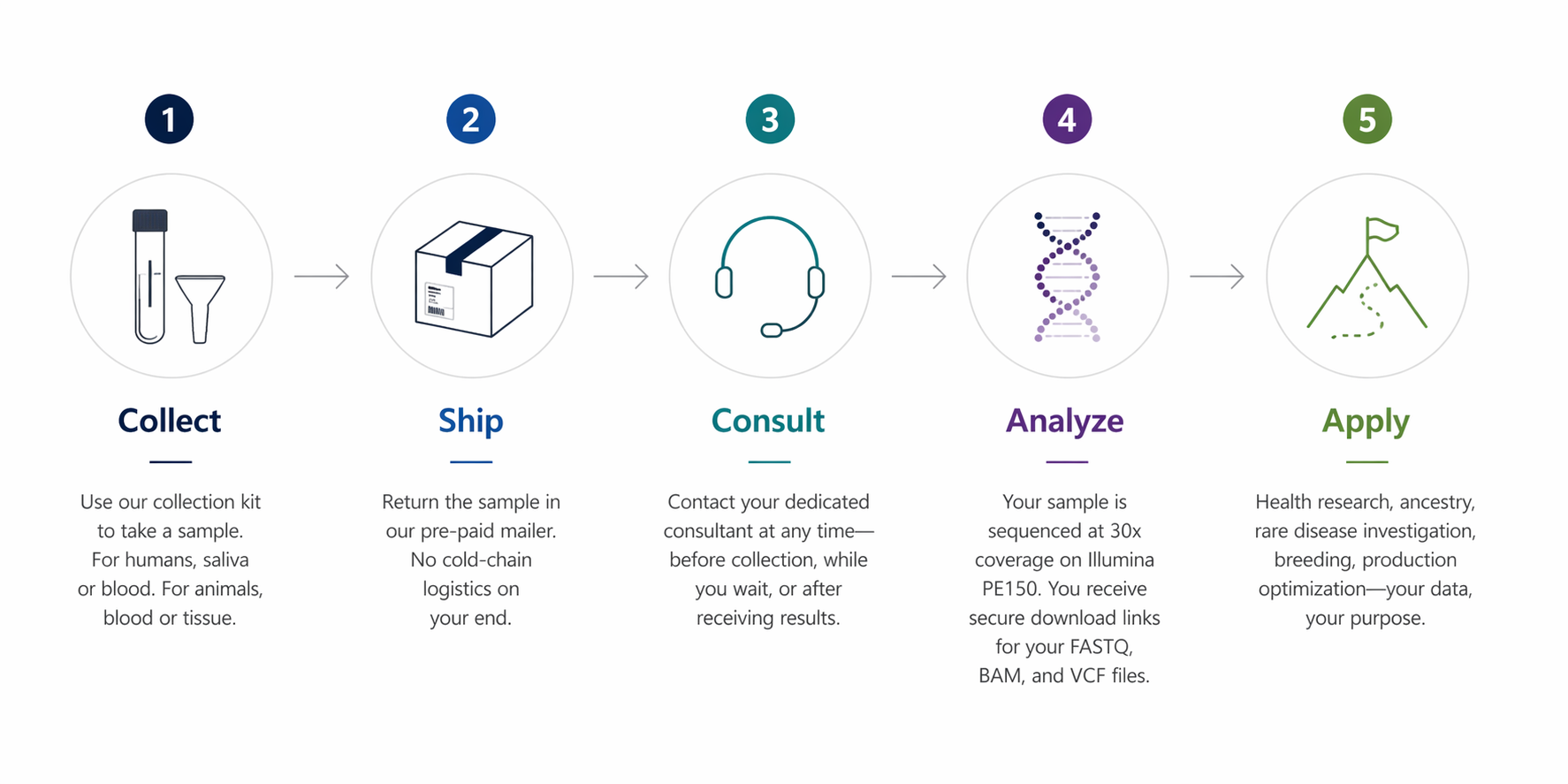
How the process works
- Collect. We send you a collection kit. For humans, this is typically a saliva or blood sample — straightforward and non-invasive. For animals, a blood or tissue sample with materials included.
- Ship. Your sample goes back in our pre-paid mailer. No cold-chain logistics to manage.
- Consult. Your dedicated consultant is available throughout the entire process. Questions before you collect, clarifications while you wait, help interpreting your results after delivery — they're there for all of it.
- Analyze. Your sample is sequenced at 30x coverage on Illumina PE150. You receive secure download links for your FASTQ, BAM, and VCF files within 2–3 weeks of sample receipt.
- Apply. Your data is yours — permanently, in open formats.
Pricing and turnaround
$599 per sample. 2–3 weeks from sample receipt.
Need results sooner? Contact us directly. Expedited processing is available.
Ready to get started?
[Sequence your genome — $599 →]
Or to speak with a consultant before you order.